Informazioni
MEXILETINA E SINDROMI MIOTONICHE
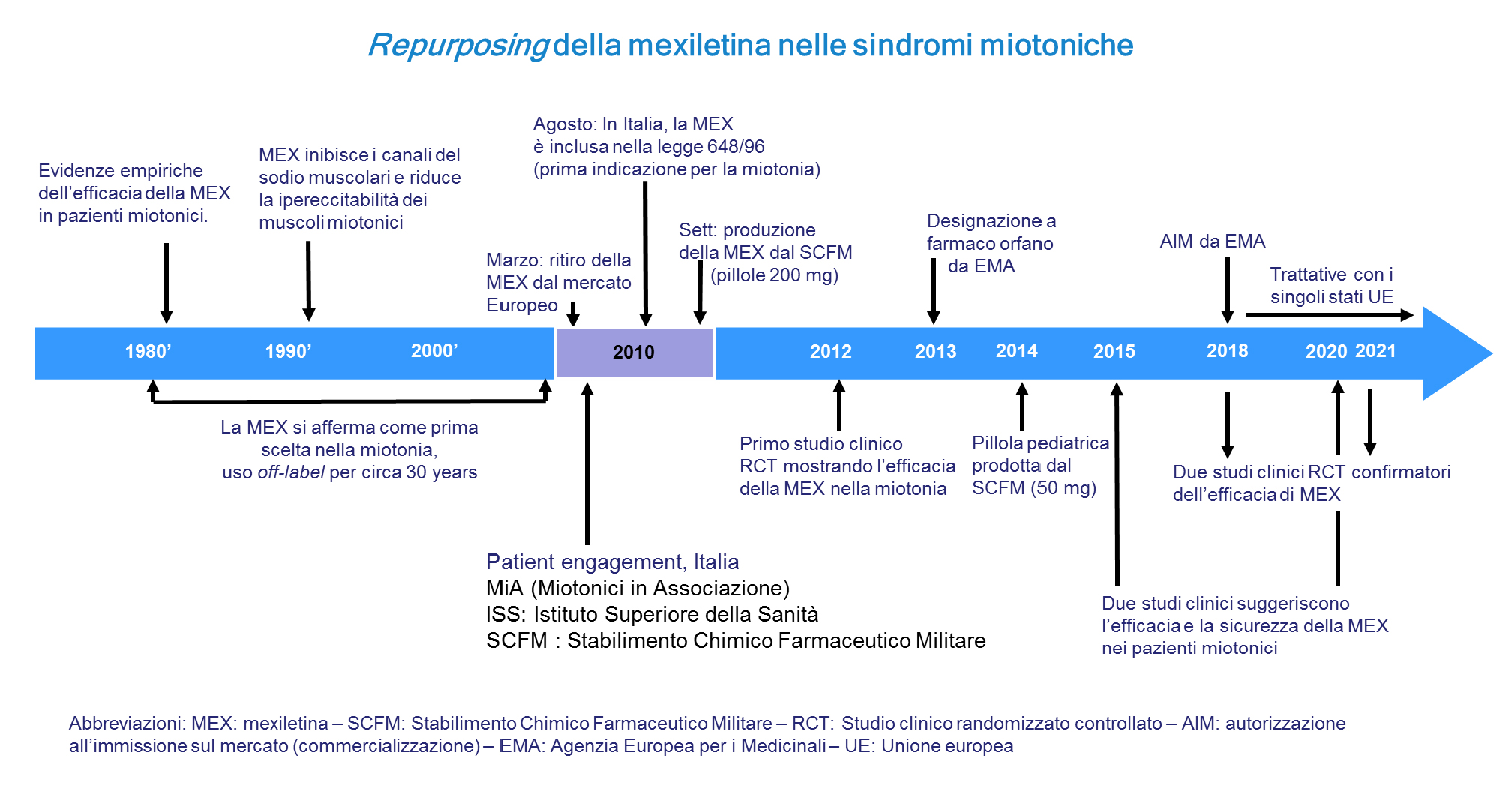
DALL’USO OFF- LABEL AL FARMACO ORFANO
Le nuove terapie delle malattie rare di origine genetica comprendono le terapie geniche e/o cellulari che, purtroppo, sono molto difficili da sviluppare e molto onerose. Nella maggior parte dei casi, quindi, la terapia farmacologica rimane l’unica soluzione per poter alleviare i sintomi, migliorare la qualità di vita e, a volte, rallentare la progressione della malattia e aumentare l’aspettativa di vita. Lo sviluppo di un nuovo farmaco è un processo lungo che può durare anche 15 anni tra l’identificazione di un candidato farmaco e la sua immissione sul mercato.
Dapprima, gli studi preclinici comprendono la selezione di una molecola promettente, la sua eventuale ottimizzazione dal punto di vista chimico e la valutazione della sua efficacia e sicurezza in modelli cellulari e/o animali. Se il farmaco supera questi test, potrà essere esaminato nell’uomo negli studi clinici in tre fasi successive (fase 1-3).
Gli studi clinici di fase 1 condotti su volontari sani mirano a verificare la sicurezza del farmaco, definire i dosaggi da usare nel paziente e capirne le proprietà farmacocinetiche, cioè i processi di assorbimento e diffusione nell’organismo, metabolismo ed eliminazione.
Gli studi di fase 2 coinvolgono un numero ristretto di malati (alcune decine) al fine di ottenere dati preliminari sull’efficacia del farmaco nell’alleviare specifici sintomi/segni della malattia. Tali dati dovranno essere confermati negli studi di fase 3 che coinvolgono centinaia o migliaia di malati. Questi studi servono anche a verificare la tollerabilità del trattamento.
Per essere validi, gli studi clinici devono essere eseguiti rispettando degli standard di qualità che garantiscano l’affidabilità statistica dei risultati. Se il farmaco mostra sufficiente efficacia e tollerabilità, il promotore chiederà l’autorizzazione all’immissione sul mercato da parte di una agenzia regolatoria, cioè l’Agenzia Europea per i Medicinali (EMA) per il mercato Europeo.
Se tutto si conclude correttamente, il farmaco sarà quindi commercializzato per una indicazione terapeutica ben precisa. Per le malattie rare, questo processo presenta evidenti ostacoli. La “rarità” dei malati preclude la condotta di studi clinici di fase 3 e non garantisce un facile ritorno economico al promotore del nuovo farmaco. Per anni, aziende farmaceutiche e altri investitori hanno “snobbato” le malattie rare perché giudicate poco redditizie. Per promuovere la ricerca nelle malattie rare, le amministrazioni pubbliche hanno quindi avviato dei programmi mirati a ridurre i costi e tempi dello sviluppo del farmaco cosiddetto orfano, dedicato al trattamento dei malati rari. Una strategia spesso utilizzata è quella del riposizionamento di un farmaco (repurposing): un farmaco già sul mercato viene usato al di fuori delle sue indicazioni (off-label) sulla base di evidenze mediche. L’uso off-label è responsabilità del medico prescrittore, meglio se viene avvalorato da un comitato etico.
Se le evidenze mediche sono sufficienti, un promotore può chiedere l’orfanizzazione del farmaco ad una agenzia regolatoria (EMA), cioè la designazione a farmaco orfano per una determinata malattia rara.
Le miotonie non-distrofiche
Le miotonie non-distrofiche (MND) sono un insieme di malattie genetiche caratterizzate da una esagerata eccitabilità delle cellule muscolari scheletriche, la quale si manifesta con un ritardo nel rilassamento muscolare dopo la contrazione e quindi rigidità del movimento. Sono malattie cosiddette rare con una prevalenza stimata a circa 1 caso ogni 100.000 individui. La rigidità muscolare può riguardare vari muscoli, compresi quelli della mano (difficoltà ad aprire il pugno), delle palpebre (difficoltà ad aprire gli occhi) e della lingua. Altri sintomi includono debolezza muscolare intermittente e dolori, e possono gravare sulla qualità di vita.
Le MND comprendono diverse malattie: la miotonia congenita è causata da mutazioni nel gene CLCN1 che codifica una proteina canale per gli ioni cloruro; le miotonie da sodio e la paramiotonia congenita sono causate da mutazioni nel gene SCN4A che codifica una proteina canale per gli ioni sodio. L’alterata funzionalità di queste proteine espresse nei muscoli scheletrici aumenta l’eccitabilità della membrana cellulare, determinando la rigidità muscolare. Tutte queste malattie hanno in comune la rigidità muscolare, ma si differenziano per i fattori scatenanti, la localizzazione all’interno dell’organismo e i sintomi secondari.
Siccome la miotonia deriva dall’ipereccitabilità della membrana dei muscoli, i farmaci che bloccano l’attività dei canali del sodio, in grado di ridurre l’eccitabilità, sono stati usati off-label, su base empirica, per trattare le miotonie. Essi comprendono farmaci antiaritmici (chinidina, tocainide e mexiletina) e farmaci anti-epilettici (fenitoina e carbamazepina).
L’effetto di questi farmaci è sintomatico; bloccando i canali muscolari, riducono l’eccitabilità della membrana e si oppongono alla rigidità muscolare, qualsisia l’origine genetica. Questi farmaci posseggono proprietà farmacologiche molto specifiche agendo solo sui canali alterati, per cui hanno pochi effetti sugli organi che presentano una normale eccitabilità (cuore e cervello), garantendo una selettività d’azione sull’organo ipereccitato (il muscolo scheletrico) e limitando il rischio di effetti avversi cardiaci e nervosi.
Dagli anni 1990, la mexiletina si è lentamente imposta come farmaco di prima scelta. Tuttavia, nel 2010, dopo un uso off-label per circa trenta anni, il farmaco è stato ritirato dal mercato europeo dall’azienda produttrice, abbandonando alla loro sorte gli utenti, i pazienti miotonici o quelli con alcune aritmie cardiache. Le ragioni di una tale decisione unilaterale non sono state divulgate. Nel corso degli ultimi 40 anni, la mexiletina è diventata il farmaco di prima scelta per le sindromi miotoniche, passando da uno status off-label a quello di farmaco orfano. Questo è stato possibile grazie all’interesse di alcuni medici neurologi e ricercatori che hanno dimostrato con metodi sempre più accurati l’efficacia del farmaco nell’alleviare la rigidità muscolare. Nell’ultimo decennio, l’associazione dei miotonici (M.I.A. ONLUS) ha portato un notevole contribuito per il riconoscimento del valore della mexiletina da parte degli enti pubblici. Gli studi clinici di elevata qualità hanno dimostrato l’efficacia, la sicurezza e la tollerabilità della mexiletina per il trattamento della miotonia. Oggi, il percorso della mexiletina viene considerato un paradigma per la scoperta di nuovi farmaci orfani per numerose malattie rare. Certo, come per qualsiasi farmaco, alcuni malati miotonici ottengono pochi benefici quando assumono la mexiletina, sia perché non giovano dell’effetto terapeutico, sia perché non tollerano il farmaco. Ma la ricerca continua per trovare farmaci alternativi.
Bibliografia/sitografia
[1] https://www.aifa.gov.it/legge-648-96
Pagina del sito web dell’Agenzia Italiana del farmaco (AIFA) che spiega la legge 640 del 1996, la quale permette l’erogazione a carico del SSN di un farmaco per una condizione diversa dalle indicazioni terapeutiche autorizzate.
[2] https://www.agenziaindustriedifesa.it/unita-produttive/stabilimento-chimico-farmaceutico-militare-firenze/
Sito web dello stabilimento chimico farmaceutico militare
[3] Statland et al., Mexiletine for symptoms and signs of myotonia in nondystrophic myotonia: a randomized controlled trial. JAMA 2012, 308:1357–1365 (doi: 10.1001/jama.2012.12607). Questa pubblicazione scientifica (in inglese) riporta i risultati del primo studio clinico (studio di tipo cross-over randomizzato e controllato) della mexiletina in 59 pazienti affetti da MND. Trattasi di uno studio internazionale che ha coinvolto anche pazienti italiani). Gli autori riportano che la rigidità muscolare lamentata dai pazienti è migliorata durante un trattamento di 4 settimane con la mexiletina rispetto al trattamento con placebo.
[4] Stunnenberg et al., Effect of mexiletine on muscle stiffness in patients with nondystrophic myotonia evaluated using aggregated N-of-1 trials. JAMA 2018, 320:2344–2353 (doi:10.1001/jama.2018.18020. Pubblicazione scientifica (in inglese) che riporta i risultati di uno studio clinico (studio di tipo N-of-1 aggregato, randomizzato e controllato) della mexiletina in 30 pazienti olandesi affetti da MND. La rigidità muscolare riportata dai pazienti risulta ridotta con la mexiletina rispetto al placebo.
[5] Vicart et al., Efficacy and safety of mexiletine in non-dystrophic myotonias: a randomised, double-blind, placebo-controlled, cross-over study. Neuromuscul Disord 2021, 31:1124–1135. (doi: 10.1016/j.nmd.2021.06.010. Pubblicazione scientifica (in inglese) che riporta i risultati di uno studio clinico (studio di tipo cross-over, randomizzato e controllato) della mexiletina in 25 pazienti francesi affetti da MND. La rigidità muscolare riportata dai pazienti risulta ridotta con la mexiletina rispetto al placebo.
[6] Pagina del sito web dell’Agenzia Europea per i Medicinali (EMA) che riporta la designazione a farmaco orfano della mexiletina per le sindromi miotoniche su richiesta dello stabilimento Chimico farmaceutico Militare (SCFM) di Firenze.
[7] Suetterlin et al., Long-term safety and efficacy of mexiletine for patients with skeletal muscle channelopathies. JAMA Neurol 2015, 72:1531–1533. (doi: 10.1001/jamaneurol.2015.2338). Pubblicazione scientifica (in inglese) che riporta i risultati di uno studio clinico retrospettivo inglese dell’uso della mexiletina a lungo termine (da 6 mesi a 18 anni) in 63 pazienti affetti da MND. La mexiletina è risultata efficace nel 60-90% dei pazienti (la % varia secondo il tipo di mutazione). Gli effetti avversi più frequentemente riportati sono stati la dispepsia, il mal di testa, le palpitazioni e la nausea. Nessun evento avverso severo è stato riportato. Attenzione, trattasi di uno studio osservazionale senza gruppo controllo (senza placebo); non permette di dimostrare una relazione causa-effetto, solo di formulare ipotesi.
[8] Modoni et al., Long-term safety and usefulness of mexiletine in a large cohort of patients affected by non-dystrophic myotonias. Front Neurol 2020, 11:300. (doi:10.3389/fneur.2020.00300). Pubblicazione scientifica (in inglese) che riporta i risultati di uno studio clinico italiano dell’uso della mexiletina a lungo termine (da 1 mese a 20 anni) in 59 pazienti affetti da MND. Solo 13 pazienti hanno interrotto il trattamento perché inefficace o intollerato o per altro motivo personale. L’effetto avverso più frequente è stato la dispepsia. Non sono stati osservati né eventi avversi severi né aritmie cardiache. Attenzione, trattasi di uno studio senza gruppo controllo (senza placebo); non permette di dimostrare una relazione causa-effetto, solo di formulare ipotesi.
Prof. Jean-Franҫois Desaphy, Farmacologo
“Università degli studi di Bari Aldo Moro”
jeanfrancois.desaphy@uniba.it