Cos’è la miotonia
Cos’è la miotonia
Per miotonia s’intende il ritardato rilassamento del muscolo dopo contrazione volontaria.
Le miotonie non distrofiche
Le miotonie non distrofiche e le paralisi periodiche sono un gruppo di malattie rare, geneticamente determinate, dovute a mutazioni di canali di membrana del muscolo scheletrico.
Le forme più frequenti sono:
- la miotonia congenita (M. di Thomsen, M. di Becker)
- la paramiotonia congenita
- la miotonia aggravata dal potassio
- le paralisi periodiche iperkaliemiche ed ipokaliemiche,
- la sindrome di Andersen-Tawil.
Dal punto di vista clinico questo gruppo di patologie si caratterizza per la presenza di “miotonia” e frequentemente di “debolezza transitoria”.
La miotonia distrofica
La distrofia miotonica, la forma più frequente di distrofia muscolare (frequenza circa 1/8000), è una patologia a carattere multisistemico, in cui oltre all’apparato muscolare scheletrico, il cui coinvolgimento è caratterizzato dalla presenza di miotonia che tipicamente si associa a debolezza ed ipotrofia dei muscoli del distretto cranico, assiali, e degli arti, sono affetti in misura variabile anche altri tessuti ed organi quali l’occhio (cataratta), il sistema nervoso centrale (disturbi cognitivi e comportamentali), il sistema endocrino (diabete mellito, distiroidismo, problemi di fertilità), la muscolatura liscia (disfagia, disturbi della motilità intestinale) ed il cuore.

UNITI
IN UN PICCOLO GESTO
IL TUO 5X1000 AIUTA LA RICERCA
C.F. 95122870637
COS’È LA MIOTONIA – Approfondimento
Per miotonia s’intende il ritardato rilassamento del muscolo dopo contrazione volontaria.
La miotonia distrofica
Di cosa si tratta?
La distrofia miotonica, la forma più frequente di distrofia muscolare (frequenza circa 1/8000) è una patologia a carattere multisistemico, in cui oltre all’apparato muscolare scheletrico, il cui coinvolgimento è caratterizzato dalla presenza di miotonia che tipicamente si associa ad una debolezza ed ipotrofia dei muscoli del distretto cranico, assiali, e degli arti, sono affetti in misura variabile anche altri tessuti ed organi quali l’occhio (cataratta),il sistema nervoso centrale (disturbi cognitivi e comportamentali) il sistema endocrino (diabete mellito, distiroidismo, problemi di fertilità), la muscolatura liscia (disfagia, disturbi della motilità intestinale) ed il cuore.
Quante forme di distrofia miotonica si conoscono?
Sono state caratterizzate due forme di distrofia miotonica: la prima, relativamente frequente, con un’incidenza di 1 caso su 10.000 nati vivi, è definita DM1 o distrofia di Steinert ed è causata dal difetto del gene della distrofia miotonica proteina kinasi (DMPK), sito sul cromosoma 19q13.3.
La seconda forma è detta DM2 o PROMM (PROximal Myotonic Myopathy), secondaria al difetto del gene della Zinc Finger Protein 9 (ZNF9), codificata dal cromosoma 3q21.
Come si trasmette la malattia?
Le distrofie miotoniche tipo 1 e 2 sono malattie ereditarie che si trasmettono con meccanismo autosomico dominante: vengono cioè colpiti indistintamente maschi e femmine e ogni figlio di una persona affetta ha un rischio del 50% di essere a sua volta colpito dalla malattia.
Qual è il difetto genetico?
In entrambi i casi è presente un’espansione patologica di alcune sequenze ripetute di DNA: un’espansione di un tratto CTG localizzata nel gene DMPK, sul cromosoma 19 è presente nei pazienti affetti da Distrofia Miotonica tipo 1 (DM1) o M. di Steinert.
Un’espansione CCTG localizzata nel primo introne del gene che codifica per ZNF9, sul cromosoma 3 è stata recentemente identificata in associazione alla DM2.
Le espansioni patologiche CTG e CCTG associate rispettivamente alla DM1 e DM2 mostrano una instabilità mitotica e meiotica, con prevalente tendenza verso l’espansione, che rendono ragione rispettivamente della variabilità di espressione clinica tra individui affetti e del fenomeno dell’anticipazione osservabile all’interno delle singole famiglie (figli di genitori affetti manifestano tipicamente i primi sintomi di malattia in un’età più giovanile rispetto all’età di esordio della malattia nei propri genitori).
Numerosi studi clinici hanno cercato di contribuire alla delucidazione dei meccanismi patogenetici della distrofia miotonica: nella DM1 l’analisi delle correlazioni fenotipo-genotipo ha documentato una correlazione tra n(CTG) nei leucociti ed età di esordio e anche tra severità del coinvolgimento muscolare ed età di esordio dei sintomi cardiologici. Non è stata invece rilevata alcuna correlazione tra n(CTG) nei leucociti ed altri segni clinici.
L’ipotesi patogenetica attualmente più accreditata per la DM1 e la DM2, supportata anche dalle loro similarità fenotipiche e molecolari, propone un effetto tossico dell’RNA trascritto dall’allele espanso, il quale a causa della sua struttura secondaria si accumulerebbe nel nucleo, con conseguente sequestro di proteine che riconoscono sequenze CUG come sito specifico di legame e regolano la trascrizione di altri geni (RNA binding proteins).
Quali le manifestazioni cliniche?
Entrambe le forme sono caratterizzate dal fenomeno miotonico cui si associa una debolezza muscolare che coinvolge, nel caso dei pazienti affetti da DM1, principalmente la muscolatura dei distretti semidistali e distali degli arti (avambraccio, mano, gamba e piede), i muscoli del volto, la muscolatura assiale. Nella DM2, invece, l’interessamento muscolare prevale nei distretti prossimali degli arti (spalle, braccia, bacino e coscia).
Tipicamente la DM1 è una malattia multi sistemica in cui sono affetti in misura variabile anche altri tessuti ed organi quali l’occhio (cataratta),il sistema nervoso centrale (disturbi cognitivi e comportamentali) il sistema endocrino (diabete mellito, distiroidismo, problemi di fertilità), la muscolatura liscia (disfagia, disturbi della motilità intestinale) ed il cuore.
Anche nella DM2 è presente un analogo interessamento multisistemico, ma con alcune differenze significative rispetto alla DM1, quali ad esempio l’assenza di rilievo di forme severe, ad espressione congenita, della malattia ed una minore severità prognostica del coinvolgimento cardiaco.
Qual è l’età di esordio dei sintomi?
L’età d’esordio è molto variabile: si conoscono, nel caso della DM1, forme congenite gravissime, forme infantili gravi e forme dell’adolescenza e dell’adulto, che sono le più comuni. Nel caso della DM2, invece, l’esordio avviene solitamente in età giovanile-adulta e non sono stati finora descritti casi congeniti di malattia.
Qual è la storia della malattia?
Nei neonati affetti dalla forma congenita di DM1 si possono presentare difficoltà respiratorie e problemi di suzione e deglutizione. Se si supera questa fase, generalmente i bambini acquisiscono le tappe motorie e manifestano un ritardo mentale e difficoltà legate alla compromissione della muscolatura distale e semidistale degli arti. Nelle forme di DM1 ad esordio giovanile-adulto, si osserva una progressiva debolezza della muscolatura scheletrica, cui si associa la miotonia, e, nel tempo, si osservano gli effetti del coinvolgimento multisistemico, con complicanze cardiache, che possono rivelarsi fatali, coinvolgimento delle funzioni cognitive, endocrinologiche, eventuale cataratta. Nel tempo si può inoltre osservare una compromissione della funzionalità della muscolatura respiratoria, con conseguenti disturbi del sonno (sindrome delle apnee ostruttive durante il sonno) e facilità alle infezioni delle vie aeree per debolezza della muscolatura respiratoria stessa.
Nel caso dei pazienti affetti da DM2, l’interessamento muscolare coinvolge principalmente la muscolatura prossimale degli arti; spesso il fenomeno miotonico è clinicamente poco evidente. È presente un interessamento multi sistemico, con possibile coinvolgimento cardiaco, endocrinologico, la comparsa di cataratta.
In entrambe le malattie possono comparire difficoltà nella deglutizione, che richiedono valutazioni specialistiche per un’opportuna caratterizzazione e trattamento.
Quali gli esami diagnostici?
Per una corretta diagnosi sono importanti innanzitutto la valutazione clinica neurologica, e l’esame obiettivo; l’esame elettromiografico e, per definitiva conferma del sospetto clinico e neurofisiologico, l’indagine genetica sul DNA estratto dai linfociti del sangue.
Nel caso in cui uno dei genitori sia affetto, si può eseguire, previa consulenza genetica specialistica, l’indagine prenatale mediante prelievo di villi coriali (frammenti di tessuto destinato a diventare placenta) alla decima-undicesima settimana di gravidanza. Tale indagine evidenzia la presenza dell’anomala espansione di nucleotidi nel feto, con risultati affidabili che permettono di dire se il figlio sarà affetto o no. Valutando poi l’entità dell’espansione, si possono avere anche indicazioni generali sulla gravità clinica. E tuttavia, poiché l’espansione è diversa nei vari tessuti, si tratta di un dato che non permette previsioni certe in questo senso.
Quale terapia?
Al presente non esiste una terapia risolutiva per la malattia.
Si ritiene necessario eseguire periodici controlli clinici e strumentali per prevenire o affrontare le complicanze a livello multisistemico. In particolare, si consigliano periodici controlli cardiologici e respiratori, al fine di prevenire complicanze potenzialmente fatali della malattia, soprattutto nel caso della DM1.
L’alimentazione va curata al fine di evitare sovraccarichi ponderali: si può intraprendere trattamento riabilitativo specifico per l’eventuale disfagia. Sono inoltre necessarie la valutazione oculistica ed endocrinologica, con un periodico profilo glicemico e controllo degli ormoni tiroidei. L’attività fisica e/o fisioterapica, purché non venga effettuata in modo attivo, cioè contro resistenza, non è controindicata in maniera assoluta, ma l’indicazione va discussa da caso a caso anche in relazione al coinvolgimento cardiaco del paziente. Infine, nei casi con compromissione della muscolatura respiratoria, può essere indicata la terapia ventilatoria di supporto.
Le miotonie non distrofiche e le paralisi periodiche
Le miotonie non distrofiche e le paralisi periodichesono un gruppo di malattie rare, geneticamente determinate dovute a mutazioni di canali di membrana del muscolo scheletrico.
Quali sono le forme note?
- la miotonia congenita (M. di Thomsen, M. di Becker)
- la paramiotonia congenita
- la miotonia aggravata dal potassio
- le paralisi periodiche iperkaliemiche ed ipokaliemiche,
- la sindrome di Andersen-Tawil.
Dal punto di vista clinico questo gruppo di patologie si caratterizza per la presenzadi “miotonia”, e frequentemente di “debolezza transitoria”.
Che cosa si intende per miotonia?
Per miotonia si intende il ritardato rilassamento del muscolo dopo contrazione volontaria. Nella vita quotidiana il paziente affetto da miotonia può presentare una difficoltà a riaprire la mano dopo aver afferrato un oggetto, ad alzarsi dalla sedia o a iniziare a camminare dopo un periodo di riposo, può incontrare difficoltà nella riapertura degli occhi dopo uno starnuto. Tale fenomeno è causato dall’alterazione della permeabilità della membrana muscolare, che diviene maggiormente eccitabile per uno squilibrio nel funzionamento di alcuni canali ionici. La miotonia tende tipicamente a ridursi fino a risolversi completamente con l’esercizio (fenomeno del warm-up), In alcuni casi invece, come ad esempio nelle miotonie da malfunzionamento del canale del sodio, il fenomeno del warm-up è preceduto da un iniziale peggioramento del fenomeno miotonico delineando il quadro della cosiddetta miotonia paradossa o paramiotonia.
Che cosa si intende per debolezza transitoria?
Si intende una paralisi flaccida di uno o più muscoli, che si può manifestare ad esempio con la difficoltà a rialzarsi da una sedia dopo un periodo di riposo o ad alzarsi dal letto fino a forme di debolezza generalizzata (la cosiddetta adinamia). Tale fenomeno è sempre transitorio ed ha una durata variabile nel tempo, da pochi secondi a ore. Spesso nelle forme in cui la debolezza transitoria è di breve durata, questa non è avvertita dal paziente come tale in quanto coincide con il fenomeno miotonico.
La debolezza transitoria si verifica per un transitorio stato di ineccitabilità della membrana muscolare, dovuta al malfunzionamento di canali di membrana. Tipicamente è più evidente nel caso delle paralisi periodiche che si manifestano proprio con attacchi di debolezza generalizzata o confinata in alcuni muscoli, scatenati solitamente dal riposo che segue uno sforzo muscolare intenso, con durata variabile da minuti a ore. Nel caso delle paralisi periodiche ipokaliemiche, che sono per la maggior parte causate da mutazioni del gene che codifica il canale del calcio, le crisi di adinamia possono invece avere una durata maggiore.
Le miotonie non distrofiche sono malattie ereditarie?
Si tratta di malattie ereditarie dovute a mutazioni di geni che codificano per canali ionici di membrana.
In particolare, la Miotonia Congenita è dovuta a mutazioni di CLCN1, il gene che codifica il canale del Cloro, sito sul cromosoma 7.
La malattia può essere ereditata come tratto autosomico dominante (M. di Thomsen), o recessivo (M. di Becker).
La miotonia di Thomsen è una malattia a trasmissione autosomica dominante: un soggetto affetto nasce cioè da un genitore malato e ha a sua volta il 50% di probabilità di trasmettere la malattia a ognuno dei figli, maschio o femmina, a seconda che trasmetta il cromosoma portatore del gene malato o quello portatore del gene sano.
La miotonia di Becker riconosce invece una trasmissione autosomica recessiva: per determinare la miotonia è cioè necessario che entrambi i cromosomi (rispettivamente quello di origine materna e quello di origine paterna) contengano il gene mutato. In tali casi quindi i genitori non risultano affetti, ma entrambi portatori della mutazione. Tale evenienza si verifica più facilmente in caso di consanguineità dei genitori. In assenza di consanguineità le probabilità aumentano nel caso in cui entrambi provengano da piccole comunità.
Le miotonie non distrofiche causate da mutazioni del gene che codifica il canale del sodio voltaggio-dipendente del muscolo scheletrico (SCN4A) sito sul cromosoma 17 si trasmettono anch’esse con modalità autosomica dominante: un soggetto affetto nasce cioè da un genitore malato e ha a sua volta il 50% di probabilità di trasmettere la malattia a ognuno dei figli, maschio o femmina, a seconda che trasmetta il cromosoma portatore del gene malato o quello portatore del gene sano.
Esse comprendono: la paramiotonia congenita, la miotonia aggravata dal potassio (includendo la miotonia fluttuante, la miotonia permanente, la miotonia responsiva all’acetazolamide), le paralisi periodiche iperkaliemiche e, più raramente, le ipokaliemiche.
La maggior parte delle forme di paralisi periodiche ipokaliemiche sono malattie autosomiche recessive dovute a mutazione del canale del calcio (CACNA1S) e non presentano il fenomeno miotonico.
La sindrome di Andersen-Tawil, infine, si manifesta con la triade sintomatologica di paralisi periodica, aritmie cardiache e malformazioni scheletriche, ed è dovuta a mutazioni del gene KCNJ2, codificante per il canale del potassio. Si trasmette con modalità autosomica recessiva.
A che età si manifestano?
Si tratta di malattie geneticamente determinate che si manifestano tipicamente in età infantile. Nel caso delle malattie da canale del Sodio l’esordio è di solito più precoce rispetto alle malattie da canale del Cloro.
C’è tuttavia da considerare che, trattandosi di un gruppo di malattie rare, quindi ancora poco conosciute, solitamente si verifica un ritardo diagnostico: infatti l’età alla diagnosi è in media giovanile – adulta.
Quali sono i sintomi?
Tipicamente i pazienti manifestano una rigidità muscolare, espressione clinica della miotonia, che nel caso delle forme da mutazione del canale del Sodio può essere talora associata a dolore. Il fenomeno miotonico si nota tipicamente ai muscoli della mano con difficoltà a rilassare il pugno, agli arti inferiori con rigidità della marcia, ma può esprimersi anche a livello dei muscoli degli occhi, con ritardo nella riapertura delle palpebre dopo chiusura forzata (ad esempio dopo uno starnuto) e talora con fugace visione doppia (per uno strabismo transitorio). L’aumentata eccitabilità della membrana muscolare con il conseguente fenomeno miotonico si può tradurre clinicamente in un’ipertrofia della muscolatura, soprattutto del collo e degli arti, conferendo ai pazienti un aspetto da culturista, nonostante non siano soggetti allenati a tal fine.
Qual è il decorso?
Trattandosi di miotonie non distrofiche, dovute a mutazione di geni che codificano per canali di membrana e pertanto definite patologie da canale di membrana, non si osserva una progressione della malattia nel tempo. Non c’è quindi una tendenza al peggioramento clinico con l’avanzare degli anni.
Quali esami sono utili per la diagnosi?
Per una corretta diagnosi sono importanti un’accurata valutazione clinica neurologica (meglio se effettuata presso un centro specializzato per la diagnosi delle malattie neuromuscolari), con la raccolta dei dati della storia familiare; quindi si procede all’esame elettromiografico, che comprende una serie di test specifici e sensibili a seconda del tipo di canale di membrana che si sospetta essere coinvolto nella singola malattia. Anche l’esame elettromiografico è bene che venga eseguito da un medico che abbia esperienza nel campo delle miotonie. Una volta confermato il sospetto clinico grazie all’esame elettromiografico, si effettuerà l’indagine genetica mirata sul DNA estratto dai linfociti del sangue, mediante un prelievo che non richiede il digiuno alimentare.
Le miotonie non distrofiche, una volta posta la corretta diagnosi genetica, sono riconosciute, nell’ambito del Sistema Sanitario Nazionale, come appartenenti al gruppo delle “Malattie Rare” per cui è previsto un codice di esenzione.
Esiste una terapia?
Al presente non esiste una terapia risolutiva per tali malattie.
Tuttavia si può intervenire con dei farmaci sintomatici che migliorano la miotonia e riducono la frequenza e l’entità degli episodi di debolezza transitoria.
Da sottolineare però che soprattutto alcuni farmaci utilizzati per ridurre la miotonia possono produrre effetti collaterali di tipo cardiologico, per cui l’opportunità di impiego e il dosaggio vanno valutati caso per caso, previa valutazione specialistica cardiologica.
La mexiletina!
Lunga vita alla mexiletina! Un futuro per i Farmaci Orfani
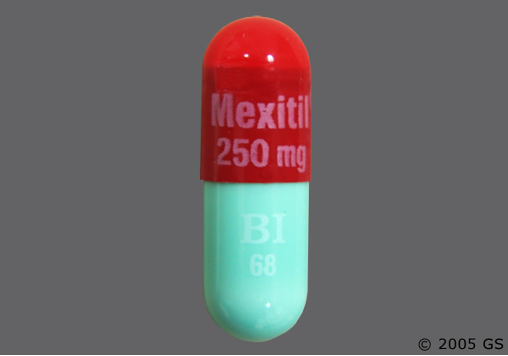
Grazie all’impegno e alla tenacia dello Stabilimento Chimico Farmaceutico Militare di Firenze, continua la produzione della mexiletina, terapia d’elezione per il trattamento della maggior parte delle sindromi miotoniche.
Vogliamo esprimere loro tutta la nostra riconoscenza.
Nel lungo iter terapeutico delle persone affette da miotonie è fondamentale l’assunzione di questo farmaco che garantisce agli adulti una vita autonoma e ai bambini un’infanzia serena.
Un farmaco – come la mexiletina – diventa come un compagno di viaggio, che consente di relativizzare l’handicap, vivendo la parte sana di noi stessi. Non è difficile immaginare cosa si è verificato quando, nel marzo 2010, ne è stata sospesa la produzione. È mancata l’aria!
Ma dopo un tempo di panico tutto è ricominciato: grazie all’impegno dello Stabilimento Chimico Farmaceutico Militare, dell’Istituto Superiore di Sanità e dell’AIFA (Agenzia Italiana del Farmaco) che hanno accolto la loro, la nostra drammaticità.
Lo Stabilimento Chimico Farmaceutico Militare fu fondato a Torino nel 1853: da allora, secondo principi etici di scienza e coscienza, garantisce la produzione di quei farmaci non più prodotti dall’industria farmaceutica. Dal 1931 lo Stabilimento opera a Firenze ed è attualmente diretto dal Generale Giocondo Santoni, al quale vanno i più sentiti ringraziamenti per la disponibilità mostrata verso le necessità espresse dalla nostra associazione, a nome dei miotonici italiani.
Una doverosa citazione ed un ringraziamento personale vanno a tutto il personale dello Stabilimento Chimico Farmaceutico Militare e al Maresciallo Camillo Borzacchiello, il principale referente per le associazioni per le malattie rare.
Da maggio 2014, la mexiletina è disponibile anche in dosaggio pediatrico.
Noi lo diciamo con forza: insieme si può! Insieme si può garantire una diagnosi precoce delle miotonie e una terapia sempre più adeguata alle esigenze di crescita dei bambini affetti da miotonia. Necessità sottolineata anche a Firenze dall’Associazione M.i.A. Onlus nel convegno del 26 giugno 2012 sui farmaci orfani organizzato dallo SCFM.
Indisponibilità della Mexiletina per pazienti miotonici
Molti miotonici sono trattati con una terapia a base di Mexitil (Mexiletina), un farmaco della categoria degli antiaritmici cardiaci, che però ha dato notevoli risultati anche come miorilassante nel caso dei pazienti affetti da miotonia.
Nelle ultime settimane si è verificata una notevole difficoltà di reperimento del farmaco nelle normali farmacie italiane.
Secondo alcune fonti mediche, la società produttrice avrebbe recentemente sospeso la produzione del Mexitil, lasciando coloro che ne facevano uso privi del farmaco e senza una valida alternativa.
La M.i.A. Onlus ha già contattato la casa farmaceutica per chiedere conferma della cessata produzione e per avere informazioni riguardo alla disponibilità residua, in particolare in Italia, alla presenza di eventuali farmaci sostitutivi, alle modalità di approvvigionamento.
È intenzione dell’associazione attivarsi per sensibilizzare il mondo scientifico sul problema e fare pressione sulle case farmaceutiche per trovare una soluzione utile per i pazienti miotonici.
L’associazione M.i.A. vuole anche porsi come punto di contatto tra i pazienti miotonici e fare da riferimento per chi si trovi alle prese con il problema di indisponibilità del farmaco.
Chi avesse incontrato queste difficoltà nel procurarsi il farmaco e volesse essere aggiornato sulle novità, può mettersi in contatto con l’associazione, tramite i nostri contatti.